
By Lorenz Adlung
Illustration by Anne-Gaëlle Goubet for SYIS
Everything is connected!
Immune cells communicate with each other thereby forming intricate interaction networks like the one on the right from our work published 2019 in Cell. With recent advances in technology, we understand more about functional interdependencies between immune cellular subsets. Immune cells reside in various tissues and circulate through the bloodstream as they are forming an interconnected web. The availability of fate-mapping mice, multiplexed cellular assays, as well as tissue profiling with unprecedented molecular and spatial resolution allow for an in-depth characterisation of social networks within the immune system.
In 2018, I speculated about integrating all this information, e.g. the transcriptional and the proteomic layer. Meanwhile, there are methods that combine several high-throughput techniques. So in theory, we can measure everything together. But how much of everything do we need to know to learn something new? The team led by Professor Bernd Bodenmiller of the University of Zurich and ETH Zurich has shown that the answer to that question depends on the tissue of interest. The team was dealing with imaging mass cytometry data – in simple terms: pointillistic pictures (see below) of tissue sections, containing information on both, RNA and protein.

The preprint from the Bodenmiller lab introduces an algorithm for intelligent experimental design planning such measurements.
Unlimited data
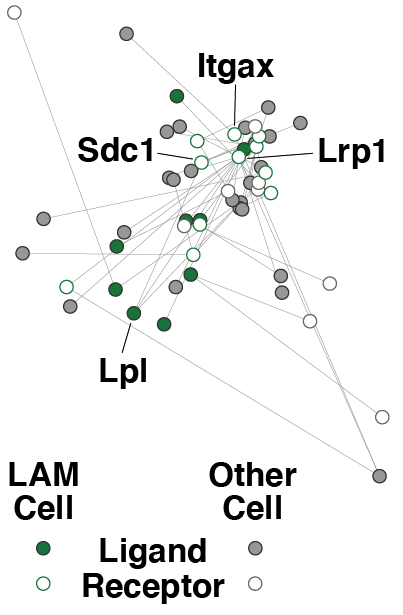
It becomes clear that data itself is no longer a limitation. It is rather our ineptitude to conceptualise ideas, which hampers the transformation (“translation”) of data into hypotheses that can be experimentally tested. I want to highlight this aspect with another example from social networks within the immune system, namely intercellular communication. Shortly after the advent of single-cell RNA-sequencing protocols, algorithms were introduced to computationally infer cellular interactions. They come in different flavours (such as “CellPhoneDB”, or “NicheNet”, to name just two) and in essence, they do all the same: they construct a bipartite graph, which is a table with two columns. The first column contains the sender cell and molecule. An example would be a pro-inflammatory macrophage expressing Il1b. The second column contains the receiver cell and molecule. The corresponding example would be a neutrophil expressing Il1r1. If the query dataset contains both sender and receiver cells and molecules, the entries are put in the same row of the bipartite graph table, and a connection can be made.
It’s a match!
Those inferred connections have to be critically evaluated for several reasons.
- It’s sparse: Single-cell RNA-sequencing data is like good Swiss cheese – full of holes. Transcriptome-wide coverage particularly for lowly expressed genes cannot be taken for granted. A way to deal with sparse data is to impute missing values. But imputed values can introduce false positives. So you assume there is a transcript whereas in reality there is none.
- It’s merely RNA: Just because there are transcripts in the data does not mean that the corresponding proteins are correctly produced and transported to the cell surface for actual interactions. Translation or post-translational modifications can be erroneous.
- Partners are unclear: For IL-1β, the receptor is well known. But since it is a complex including also IL1-R3, presence of IL-1β and IL1-R1 alone will not suffice for an interaction. There are other examples for which interacting pairs of ligand and receptor are not yet clearly defined. The triggering receptor expressed on myeloid cells 2 (Trem2) is such an example – it is a lipid receptor, but the exact molecules, which are sensed, remain elusive.
- Proximity unknown: Sender cells secrete molecules that can be bound by receptor molecules on the surface of receiver cells. However, such an interaction only occurs if they are in proximity allowing a physical interaction of both entities prior to eventual depletion.
From my experience, the detection of “significant” cellular interactions with the methods mentioned above depends on data quality and data processing. If I fail to identify specific subsets of immune cells within my data, the chance is very high that the algorithms miss interactions among those subsets. The reason is that specific (and therefore rare) interactions are “diluted” below the detection limit in the coarser (and therefore bigger) immune populations.
Keep it simple!
Accordingly, an ultimate proof of inferred interactions is in vitro. After all those new-gen technologies, we are going back to old-school co-cultivation experiments. Take macrophages and/or neutrophils from conditional knock-out mice, put them together and test whether cell communication is abrogated as expected! I believe that such straightforward experiments are a precondition to probe the existence and functional relevance of social networks within the immune system.
Post scriptum
Data-driven predictions go hand in hand with experimental validation. If you share my network perception of the immune system and beyond, you may want to contribute to our special issue of Cells in the context of the gastro-intestinal tract:
http://www.mdpi.com/journal/cells/special_issues/Cells_ColoNet
The SYIS does not guarantee the accuracy of the content published in this blog. The content does not necessarily reflect the opinion or views of the SYIS.

Leave a comment